

Investors interested in RedHill Biopharma Ltd. (RDHL) are keeping a close watch on three pending catalysts. These include the initiation of the confirmatory Phase 3 program with RHB-105, a potential new standard-of-care for H. pylori infection, interim DSMB analysis from the Phase 3 study with RHB-104, a potential game-changer for the treatment of Crohn’s disease, and top-line results from the Phase 3 acute gastroenteritis and gastritis study with Bekinda®, a novel extended-release formulation of ondansetron. These three Phase 3 programs make up the core of RedHill’s focus on GI-related disorders, and alone make the company an attractive late-stage specialty pharma story.
In the background lay two Phase 2 novel, orally-administered small molecule oncology drug candidates, Yeliva™ and Mesupron. My delve into Yeliva™, a program that continues to receive significant U.S. National Cancer Institute (NCI) support, found a novel drug with a validated mechanism and potential to address multiple oncology and inflammatory indications. Recent positive Phase 1 data with Yeliva™ in patients with advanced solid tumors sets the stage for RedHill’s expansion into oncology. For this article, I look at Mesupron, another intriguing mid-stage oncology candidate with a novel mechanism for potentially multiple solid tumor indications. To date, Mesupron has been studied in ten clinical trials, including eight Phase 1 trials and two Phase 2 trials.
Mesupron (WX-671, upamostat) is a Phase 2, proprietary, first-in-class, synthetic urokinase-type plasminogen activator (uPA) inhibitor. The drug, which is an oral amidoxime-prodrug of the active metabolite WX-UK1, acts as a serine protease inhibitor interfering with the uPA system. The uPA system plays an important role in invasion and metastasis of solid tumors (1), as well as fibrinolysis, cell migration, angiogenesis, wound healing, embryogenesis, and tumor cell dissemination (2). Overexpression of uPA and/or its receptor (uPAR) is one of the critical components of tissue metastasis and strongly correlated with poor prognosis and disease recurrence (3, 4). Researchers consider this biomarker of high clinical significance (5, 6).
Importantly, however, the uPA system is not essential for survival under physiologically healthy conditions (7). As such, this creates the ideal therapeutic target for patients with malignant solid tumors because elevated levels of uPA can be effectively knocked down without severe cytotoxic effects to the patient. It also presents the opportunity to test uPA inhibitors in patients with solid tumors who are unlikely to benefit from or resistant to adjuvant chemotherapy (8, 9).
– A Validated Target
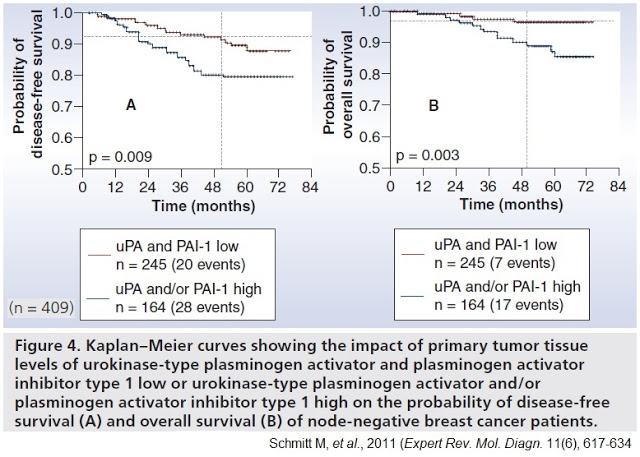
Further rationale for the targeting of uPA was demonstrated by the landmark Chemo-N0 clinical study (NCT01317108). Chemo-N0, sponsored by Universitätsklinikum Hamburg-Eppendorf between 1993 and 1998, was the first prospective randomized multicenter trial designed to evaluate the clinical utility of uPA/PAI-1 prospectively. The 698-patient trial enrolled patients with high-risk lymph node-negative breast cancer, randomized to chemotherapy versus observation and stratified by uPA/PAI-1 expression levels.
The trial was designed to answer two important questions: 1) Can the reported prognostic impact of uPA and PAI-1 be validated in a prospective multicenter therapy trial? Does low uPA/PAI-1 identify those low-risk N0 patients who are candidates for being spared necessity and burden of adjuvant chemotherapy? And, 2) Do uPA/PAI-1 high-risk patients benefit from adjuvant CMF chemotherapy?
Results from the trial demonstrated the clinical importance of uPA to the highest level of evidence (LOE-1) according to the principles of the Tumor Marker Utility Grading System (10). Importantly, in the 245 patients with low uPA/PAI-1 tumor levels, researchers observed only 20 disease recurrences, corresponding to a disease recurrence probability of 10.5% within five years. Patients with elevated uPA and/or PAI-1 values presented with a substantially higher probability of 5-year disease recurrence at 20.6%. The statistical outcome was also applicable to the overall survival of the patients.
– Early Clinical History
The clinical experience to date with Mesupron or its intravenous precursor, WX-UK1, is extensive. WX-UK1 originally developed by Wilex AG, a German biopharma company spin-off of the Department of Obstetrics and Gynecology of the Technical University of Munich. Four Phase 1 studies were conducted with intravenous WX-UK1 (see below). No serious adverse events related to WX-UK1 administration were observed, and no safety issues in capecitabine combination studies were noted. The combination of WX-UK1 with capecitabine showed encouraging effects in cancer patients, including evidence of prolonged stable disease and partial responses, and thus clinical benefit (11).
Wilex AG then developed an oral prodrug, upamostat, branded as Mesupron, for the next four Phase 1 clinical studies. The oral formulation showed excellent bioavailability and pharmacokinetics, with no serious adverse events or dose-limiting toxicities that would impact therapeutic use in cancer patients (12).

– Phase 2 Clinical Trials
Wilex AG completed two sizable Phase 2 clinical trials with Mesupron. Data from both studies was presented at the American Society of Clinical Oncology (ASCO) meeting in May 2010.
Pancreatic Cancer: This Phase 2 trial (NCT00499265) enrolled a total of 95 patients with locally advanced, non-resectable, non-metastatic pancreatic cancer randomized to three cohorts receiving daily oral Mesupron 200 mg, 400 mg, or placebo. All patients were treated with weekly gemcitabine (1,000 mg/m2 i.v. as per SPC). Treatment continued until disease progression or toxicity. Safety was assessed by measuring vital signs, laboratory parameters (hematology, blood chemistry, coagulation) and ECGs. Efficacy endpoints were response rate at the various staging intervals, progression free survival, time to first metastasis, overall survival as well as changes in tumor- and uPA-system-related markers. All 95 patients were accrued between June 2007 and Aug 2008. Efficacy was assessed by a central reader at regular intervals based on digital CT images.
By the end of 2009, two patients were still on treatment without signs of progression, 64 patients had died. Preliminary analysis of overall survival showed an increase in overall survival from 10.2 months (gemcitabine alone) to 13.5 months for the combination of gemcitabine and Mesupron. One year survival increased from 37% with gemcitabine to 53% when combined with 400 mg Mesupron (13). These are impressive results, although the trial was not powered to statistical significance.
Breast Cancer: This Phase 2 trial (NCT00615940) enrolled 132 female patients with HER2-receptor negative metastatic breast cancer randomized to receive Mesupron (200 mg orally daily for 21 days) plus capecitabine (1,000 mg/m2 orally twice daily for 14 days) vs. capecitabine (1,000 mg/m2 orally twice daily for 14 days) in 3 week treatment cycles until progressive disease or unacceptable toxicity. The primary endpoint was the efficacy of the combination of WX-671 plus capecitabine compared to capecitabine alone as assessed by comparison of progression free survival (PFS). The secondary objectives were overall survival (OS), objective response rate (ORR), safety, and tolerability, and to assess the pharmacokinetics of Mesupron and capecitabine when combined (14).
Administration of Mesupron and capecitabine resulted in median PFS values of 8.3 months (95% confidence interval (CI) 5.6 to 9.6) in the total study population and 7.5 months (95% CI 4.2 to 12.8) in the control group given only capecitabine. However, in the group of patients who had received prior adjuvant chemotherapy, PFS increased from 4.3 months (95% CI 2.6 to 9.7) in those treated with capecitabine alone to 8.3 months (95% CI 5.6 to 10.9) in the group receiving Mesupron and capecitabine. In addition, objective response rate was higher in the group receiving the combination therapy compared with those receiving capecitabine alone (20% versus 12% at week 24). Importantly, the combination of Mesupron and capecitabine was reported to be safe and well tolerated, with no unexpected toxicities other than those attributable to capecitabine (15).

The results of the above Phase 2 trials confirm earlier work that uPA is a marker for aggressive cancer and an effective target for therapeutic intervention (16). In the pancreatic cancer trial, Mesupron 400 mg provided a 49% improvement in overall survival at 12 months compared to gemcitabine (Gemzar®) alone. In Phase 2 breast cancer study, Mesupron 200 mg increased objective response rate and median progression-free survival in the subgroup of patients who had received adjuvant chemotherapy (aggressive primary disease) and provided an improvement in overall response rate compared to capecitabine (Xeloda®) alone. Although neither Phase 2 trial was powered for statistical significance, the data shows clear proof-of-concept and provides a platform for advancement into larger, well-powered studies in patients with advanced disease.
What’s Next For Mesupron?
In June 2014, RedHill acquired the exclusive worldwide rights to Mesupron, excluding China, Hong Kong, Taiwan and Macao, for all indications (17). RedHill paid Wilex AG $1 million (USD), and will pay tiered royalties on net revenues ranging from mid-teens to 30% once commercialized. RedHill is responsible for all development, regulatory, and commercialization of Mesupron. Since that time, RedHill has been conducting additional non-clinical studies with the drug to understand further the molecular target and better define the patient population. I suspect, based on the promising Phase 2 overall survival data in pancreatic cancer that this will be the primary focus on management with Mesupron in 2017; however, the progression-free survival data in post-adjuvant metastatic breast cancer is also promising and makes an excellent expansion opportunity for the company.
Conclusion
RedHill Biopharma recently reported financial results for the second quarter ending June 30, 2016. The company exited the quarter with $47.7 million in cash and investments (18), sufficient to fund operations and drive forward the company’s late-stage gastrointestinal and inflammation-based pipeline. As noted above, there are some very important catalysts on the horizon, including a DSMB interim look at the Phase 3 MAP-US trial with RHB-104 for Crohn’s disease, as well as final data from the Phase 2a exploratory trial with RHB-104 in multiple sclerosis. Phase 3 data from Bekinda® in gastroenteritis is expected before the end of the year; and, the confirmatory Phase 3 RHB-105 trial in patients with active H. pylori infection is expected to be initiated in the coming months.These important catalysts certainly make RedHill a name to watch over the next few months; moreover, earlier-stage assets in the pipeline, including Yeliva™™ and Mesupron, are also interesting and present RedHill the opportunity to expand its business into oncology. Mesupron targets a validated cancer biomarker of high significance and Phase 2 data points to clear utility in both pancreatic and metastatic breast cancer. These assets provide diversification and upside to the story as RedHill enters a pivotal period over the next year.